Primary amenorrhoea is defined as the absence of menstruation by the age of 15 years in females with normal secondary sexual development or by the age of 13 years in those who lack signs of pubertal development.1 The diagnosis of primary amenorrhoea involves a thorough physical examination and laboratory tests including hormone assays and genetic testing, and imaging studies. A large list of causes has been postulated, and subcategorized into hypogonadotropic hypogonadism (HH), hypergonadotropic hypogonadism, and normogonadotropic hypogonadism.2 Among the causes of HH are constitutional delay of growth and puberty, congenital HH, functional hypothalamic amenorrhoea, and pituitary tumors. Isolated HH (IHH) is uncommon and is typically classified as either normosmic (having no smell defect) or anosmic (Kallmann syndrome).2
Case Report
A 22-year-old Omani woman presented with a history of primary amenorrhoea. She had never experienced menstrual bleeding or cyclical pain but had noticed changes in her breast and pubic and axillary hair growth along with a sudden increase in height at 17 years of age. She denied any headache, blurring of vision, anosmia, galactorrhea, constipation, or weight loss. She had no history of proximal muscle weakness, easy bruising, or purplish striae. She denied any history of excessive exercise or strict dieting to lose weight. Apart from allergic rhinitis, her medical history was unremarkable and she was not on any medication. Her antenatal and birth history was uneventful. Her parents are consanguineous with no family history of similar illnesses.
On examination, the patient appeared well-nourished with a body mass index of 22 kg/m2 (weight = 59.5 kg; height = 165 cm). Her breast, axillary, and pubic hair development was at Tanner stage 2. There were no abnormalities noted on abdominal, pelvic, or neurological examination. The baseline blood test results (full blood count, thyroid function tests, and prolactin levels) were within the normal range. Hormone profiles were suggestive of HH: oestradiol < 0.02 nmol/L (reference: 0.63–1.15), follicle-stimulating hormone (FSH): 0.6 IU/L (3.5–12.5), and luteinizing hormone (LH): 0.1 IU/L (2.4–12.6). Other pituitary hormone profiles were within their normal ranges.
Karyotyping revealed a normal female karyotype (46,XX). A pelvic ultrasound showed an atrophic uterus with normal ovaries [Figure 1]. Brain magnetic resonance imaging revealed a hypoplastic pituitary gland, but an otherwise normal brain structure [Figure 2].
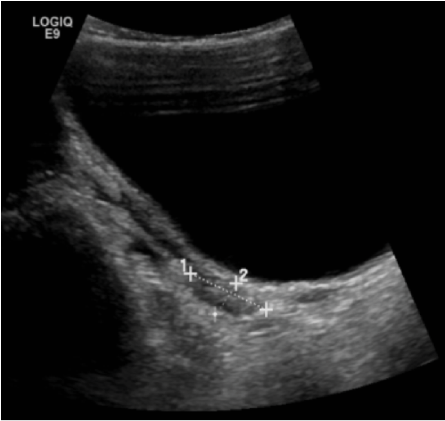 Figure 1: Ultrasound image showing atrophic uterus sized 2.5 × 1.1 cm.
Figure 1: Ultrasound image showing atrophic uterus sized 2.5 × 1.1 cm.
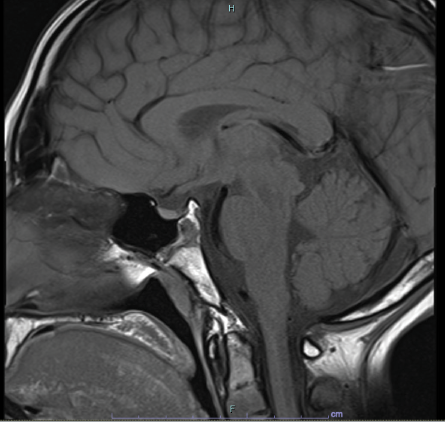 Figure 2: Brain MRI indicating hypoplastic pituitary gland.
Figure 2: Brain MRI indicating hypoplastic pituitary gland.
The patient’s atrophic uterus, normal karyotyping, and the unexpected finding of HH enabled us to identify two primary differential diagnoses: CHH or CDGP. Both CHH and CDGP are diagnosed by excluding other differential diagnoses and differentiating between the two is often difficult. In the current case, some clues favored CHH as the likely diagnosis, such as the normal bone age and genetic testing results. Genetic testing revealed a diagnosis of autosomal recessive HH 8 with or without anosmia (KISS1R, NM_032551.4:c.443T>C; p.Leu148Ser). The patient’s olfactory sensations were normal, and Kallmann syndrome was ruled out.
The patient was started on combined contraceptive pills (drospirenone 3 mg; ethinyl oestradiol 0.03 mg) for six cycles. As her bone density was subpar for her age and sex (Z score for lumbar spine: -3.6; hip: -1.8), vitamin D and calcium supplements were also prescribed. She started to have regular cycles after the combined contraceptive pills were started. The follow-up pelvis ultrasound after six months showed an interval increase in the size of the uterus compared to the first ultrasound (size = 5 × 1.5 cm, vs. 2.5 × 1.1 cm at baseline) [Figure 3]. She was advised to continue taking the combined contraceptive pills with regular follow-up pelvic imaging every six months.
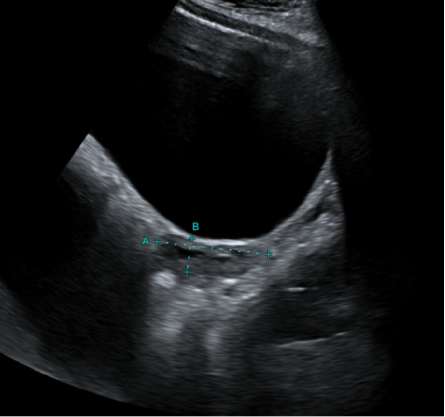 Figure 3: Ultrasound image taken after six months of hormone therapy shows a growing uterus of size
Figure 3: Ultrasound image taken after six months of hormone therapy shows a growing uterus of size
5 × 1.5 cm.
Discussion
Diagnosing primary amenorrhoea requires history taking, clinical examination, laboratory testing, and imaging. Our patient presented with primary amenorrhoea, HH, an atrophic uterus with normal ovaries, and a hypoplastic pituitary gland. Genetic testing confirmed the diagnosis of autosomal recessive HH 8, with or without anosmia.
CHH is a rare disorder with an estimated prevalence of about 1 in 8000 individuals. It is caused by abnormalities in episodic gonadotropin-releasing hormone (GnRH) secretion, causing disruptions in the normal development of reproductive function.2 Any dysfunction in GnRH synthesis, mechanism of action, or both can lead to HH. The causes of CHH could be attributed to (a) variations in the formation or shift of GnRH neurons during prenatal stages (e.g., Kallmann syndrome) or (b) structural abnormalities that specifically impact GnRH formation, maturation, or signaling in the hypothalamus, without altering the typical anatomical location of GnRH neurons. Patients in the latter group do not experience neurological disorders and possess a normal sense of smell, indicating normosmic CHH.3,4
IHH is an uncommon condition that leads to the decreased or absent production of GnRH which in turn reduces LH and FSH, ultimately lowering the levels of sex hormones.5 IHH may occur due to various genetic mutations that affect the production or action of GnRH, LH, or FSH, including mutations in genes such as KISS1R, GNRHR, TAC3, and TACR3. IHH can also occur due to non-genetic factors such as tumors, brain injuries, or infections that affect the hypothalamus or pituitary gland.5
Mutations in the KISS1R gene are rare and can disrupt the normal function of the kisspeptin receptor, leading to reduced or absent GnRH secretion and subsequent HH.6 The specific mutation c.443T>C (p.Leu148Ser) is rare, and has been reported only in a few studies.7–10 In a French study that conducted a genetic analysis of 603 patients (399 men and 204 women) with normosmic congenital HH, 12 (2%) had at least one mutation, and only one patient had the c.443T>C (p.Leu148Ser) mutation.7 Meanwhile, a Saudi Arabian study reported six members from one family who were homozygous for c.443T>C(p.Leu148Ser) and had HH.7–9
Symptoms of IHH typically include delayed or absent puberty, infertility, and low levels of sex hormones. In some cases, individuals with IHH may also experience other health problems related to low sex hormone levels, such as osteoporosis or cardiovascular disease. Treatment for IHH typically involves hormone replacement therapy.5 Genetic testing is recommended for individuals with a familial history of HH. Genetic counseling is also recommended for individuals and families who have been diagnosed with HH or are at risk for the condition.
In individuals with primary hypogonadism, estrogen replacement therapy has been observed to stimulate uterine growth. A case study has been reported involving a 23-year-old woman with 46,XX gonadal dysgenesis and primary amenorrhoea. The patient was administered oral conjugated equine estrogen daily as a hormone substitution therapy. Six months later, she exhibited Tanner stage III, and within 18 months, Tanner stage V. A repeat pelvic ultrasound unexpectedly revealed rudimentary uterine buds measuring 1.3 × 3.8 cm, but no ovaries or upper part of the vagina were observed. At 24 months, a pelvic magnetic resonance imaging confirmed a developing uterus.10
A retrospective study from Korea reported the effect of two years of estrogen replacement therapy on uterine development in a 35 young women with primary hypogonadism and primary amenorrhoea.11 They were categorized into three groups based on the cause of hypogonadism: (a) Turner syndrome, (b) HH following brain surgery, and (c) premature ovarian insufficiency following cancer treatment. After estrogen replacement therapy, pelvic ultrasound showed a significant increase in the mean uterine cross-sectional area, but the group with premature ovarian insufficiency had a significantly smaller final uterine cross-sectional area than the other two groups. Logistic regression analysis also revealed that etiology and cumulative estrogen dose were associated with uterine maturation.11
Conclusion
Primary amenorrhoea is a rare condition that requires a thorough diagnostic evaluation. This case report highlights the importance of a systematic approach in evaluating of primary amenorrhoea, including detailed history taking, clinical examination, laboratory tests, imaging, and genetic testing.
Disclosure
The authors declare no conflicts of interest. Informed written consent was obtained from the patient.
references
- 1. ACOG Committee Opinion No. ACOG committee opinion no. 651: menstruation in girls and adolescents: using the menstrual cycle as a vital sign. Obstet Gynecol 2015 Dec;126(6):e143-e146.
- 2. Seppä S, Kuiri-Hänninen T, Holopainen E, Voutilainen R. Management of endocrine disease: diagnosis and management of primary amenorrhea and female delayed puberty. Eur J Endocrinol 2021 May;184(6):R225-R242.
- 3. Francou B, Paul C, Amazit L, Cartes A, Bouvattier C, Albarel F, et al. Prevalence of KISS1 receptor mutations in a series of 603 patients with normosmic congenital hypogonadotrophic hypogonadism and characterization of novel mutations: a single-centre study. Hum Reprod 2016 Jun;31(6):1363-1374.
- 4. Quinton R, Duke VM, Robertson A, Kirk JM, Matfin G, de Zoysa PA, et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf) 2001 Aug;55(2):163-174.
- 5. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, et al. Expert consensus document: European consensus statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2015 Sep;11(9):547-564.
- 6. Seminara SB, Hayes FJ, Crowley WF Jr. Hypogonadotropic hypogonadism. N Engl J Med 1998;338(7):417-422.
- 7. Pallais JC, Bo-Abbas Y, Pitteloud N, Crowley WF Jr, Seminara SB. Neuroendocrine, gonadal, placental, and obstetric phenotypes in patients with IHH and mutations in the G-protein coupled receptor, GPR54. Mol Cell Endocrinol 2006 Jul;254-255:70-77.
- 8. Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS Jr, Shagoury JK, et al. The GPR54 gene as a regulator of puberty. N Engl J Med 2003 Oct;349(17):1614-1627.
- 9. Bo-Abbas Y, Acierno JS Jr, Shagoury JK, Crowley WF Jr, Seminara SB. Autosomal recessive idiopathic hypogonadotropic hypogonadism: genetic analysis excludes mutations in the gonadotropin-releasing hormone (GnRH) and GnRH receptor genes. J Clin Endocrinol Metab 2003 Jun;88(6):2730-2737.
- 10. Thewjitcharoen Y, Veerasomboonsin V, Nakasatien S, Krittiyawong S, Himathongkam T. Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal dysgenesis: a missed opportunity for prevention of osteoporosis. Endocrinol Diabetes Metab Case Rep 2019 Dec;2019:19-0122.
- 11. Kim HJ, Lee DY, Yoon BK, Choi DS. Uterine development after estrogen replacement therapy in women with different etiologies of primary hypogonadism. Obstet Gynecol Sci 2016;59(4):315-321.