Karyomegalic interstitial nephritis (KIN) is a rare inherited kidney disease that is frequently present in the second decade of life with hematoproteinuria, recurring respiratory infections, and progressive chronic kidney disease (CKD), leading to end-stage kidney disease before 50 years of age.1 There is no obvious sex or ethnic bias. Renal biopsy commonly reveals karyomegalic cells, which can also be found in the liver, lungs, skin, gastrointestinal tract, heart, and brain. KIN is associated with chronic tubulointerstitial nephritis with expansion of tubular nuclei on electron microscopy.2
Autosomal recessive polycystic kidney disease (ARPKD) is one of the most prevalent inherited polycystic kidney diseases (PKDs) in infants and children with an estimated incidence of 1:20 000 to 1:40 000 live births, and predictably higher in isolated or inbred populations.3–5
Here, we present the case of a pediatric patient with a homozygous polycystic kidney and hepatic disease 1 (PKHD1) variant causing ARPKD, and a concurrent homozygous Fanconi anemia-associated nuclease 1 (FAN1) variant causing KIN. To our knowledge, this is the first report of these two rare causes of chronic kidney disease found in a single patient. Due to multiple consanguinity, the PKHD1 homozygous allele was also present in the proband’s father, mimicking an autosomal dominant pattern of inheritance of PKD.
Case Report
A four-month-old boy was referred to our tertiary referral hospital. He was born at 35 weeks gestation weighing 1.92 kg. Antenatally, he had intrauterine growth restriction and large echogenic kidneys with oligohydramnios.
An abdominal ultrasound scan showed bilaterally enlarged kidneys (right = 8.2 cm, left = 7.2 cm) with an increase in cortical echogenicity, hypoechoic pyramids with cystic changes, and echogenic calcific areas [Figure 1]. He was noted to have hypertension, which was treated with propranolol. His kidney function was initially normal but gradually deteriorated over time reaching CKD stage III by three years of age. Urine dipsticks showed proteinuria and hematuria. The urine protein creatinine ratio was 99.7 mg/mmol (reference range < 20 mg/mmol). He had an enlarged liver (3 cm below the costal margin), but liver function tests showed normal results.
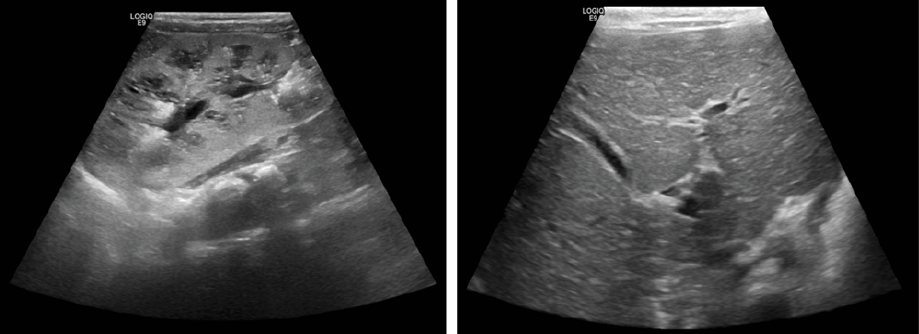 Figure 1: The abdominal ultrasound results of the proband. Kidney ultrasonography showing enlarged kidneys (right kidney 8.2 cm in length) (left panel). Liver ultrasonography showing mild dilatation of intrahepatic ducts (duct diameter 3 mm) but no cystic dilatation of the biliary tree (right panel).
Figure 1: The abdominal ultrasound results of the proband. Kidney ultrasonography showing enlarged kidneys (right kidney 8.2 cm in length) (left panel). Liver ultrasonography showing mild dilatation of intrahepatic ducts (duct diameter 3 mm) but no cystic dilatation of the biliary tree (right panel).
The patient’s parents were first cousins, and his father had PKD from childhood, which had now advanced to CKD stage II. An uncle also had PKD. The patient’s younger brother when six months old had also presented with failure to thrive, anemia, and hypertension. His abdomen ultrasound scan showed soft, palpable bilateral enlarged kidneys with multiple cysts and an enlarged liver (4 cm below the costal margin).
Whole exome sequencing (WES) in the proband (our patient) identified a homozygous likely pathogenic variant in FAN1(NM_014967.4: c.2854C>T, p.R952*) in addition to a homozygous nonsense variant of uncertain significance in PKHD1 (NM_138694.3: c.406A>G, p.T136A) [Figure 2]. Both variants were confirmed with bi-directional Sanger sequencing, and family segregation analysis was performed. Familial carrier testing in parents and the affected sibling confirmed segregation of the PKHD1 variant in a homozygous state in the patient’s father and brother and in a heterozygous state in the mother. It also confirmed that both his parents and brother were heterozygous carriers of the FAN1 (NM_014967.4:c.2854C>T, p.R952*) variant.
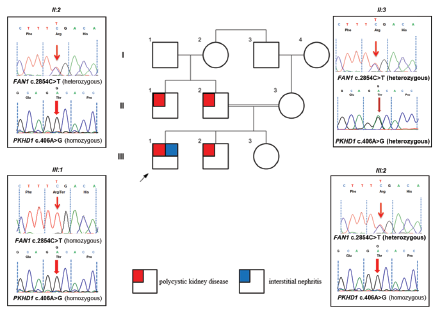 Figure 2: Pedigree diagram and Sanger sequencing chromatograms. A heterozygous nonsense FAN1 c.2854C>T, p.(R952*) change in father (II:2) and a homozygous FAN1 c.2854C>T, p.(R952*) in the affected proband (III:1) seen, which may have led to interstitial nephritis. Familial segregation of the PKHD1 missense variant c.406A>G, p.T136A, which is homozygous in father (II:2) and proband (III:1) and his sibling (III:2), all with polycystic kidney disease phenotypes.
Figure 2: Pedigree diagram and Sanger sequencing chromatograms. A heterozygous nonsense FAN1 c.2854C>T, p.(R952*) change in father (II:2) and a homozygous FAN1 c.2854C>T, p.(R952*) in the affected proband (III:1) seen, which may have led to interstitial nephritis. Familial segregation of the PKHD1 missense variant c.406A>G, p.T136A, which is homozygous in father (II:2) and proband (III:1) and his sibling (III:2), all with polycystic kidney disease phenotypes.
A diagnosis of ARPKD secondary to the PKHD1 pathogenic variant with concurrent KIN secondary to a pathogenic FAN1 variant was made. The proband, father, and sibling are currently being managed for progressive CKD.
Discussion
KIN is a rare genetic renal disease that has an autosomal recessive mode of inheritance, where an association between mutations in the FAN1 gene and KIN was recently made.6 FAN1 is located on chromosome 15 and encodes a DNA endo- and exonuclease, which acts to repair DNA, a key step in the Fanconi anemia DNA damage response pathway.6
Our patient was found to be homozygous for a nonsense variant in FAN1 which produced a premature stop codon at position 2856. This variant has been reported in the Human Genome Mutation Database (HGMD ID: CM158612) and was previously reported as a germline mutation causing hereditary colorectal cancer.7 The same variant had previously been submitted to the ClinVar database (ID: 24413339) as a germline mutation associated with KIN.8 There is no specific treatment for KIN at present but genetic counseling for affected families should be considered. KIN has been reported recently in conjugation with leukocyte chemotactic factor 2 amyloidosis, the third most common cause of amyloid nephropathy presenting with CKD.9
The classic clinical presentation of ARPKD is characterized by bilaterally enlarged kidneys with multiple cysts mostly developing in distal tubules and collecting ducts. Congenital hepatic fibrosis due to ductal plate malformation is another typical feature of ARPKD.4
In this consanguineous family, the pattern of PKD that presented in the adolescence of the father and uncle mimicked autosomal dominant PKD, highlighting the importance of obtaining a molecular diagnosis of cystic kidney diseases due to phenotypic overlaps. A 1998 study reported 13 members of a consanguineous family with different features of Alport syndrome. They carried homozygous or compound heterozygous splicing variants in COL4A3, creating a pseudodominant transmission pattern.10
In another study, two inherited kidney disorders were reported in a patient with both ADPKD and Alport syndrome.11 The coexistence of such severe, inherited kidney disorders is very rare, illustrating the significance of considering WES as a method of choice for genetic diagnosis in the setting of a positive family history for a hereditary disorder.
Conclusion
We have presented a case in which two rare genetic causes of CKD (KIN and ARPKD) were present within a highly consanguineous Omani family. This case highlighted the critical level of a homozygosity underlying inherited kidney disease in the Omani population and the importance of undertaking precision molecular diagnosis to guide treatment. Genetic counseling is also necessary for such families. We recommend WES as a routine genetic diagnostic tool for children with CKD, especially in consanguineous families.
Disclosure
The authors declare no conflicts of interest. Informed consent was obtained from the patient's father. The case was referred for WES through the MOH genetic referral committee (Royal Hospital, Oman). John A. Sayer is funded by Kidney Research UK and the Northern Counties Kidney Research Fund.
Acknowledgments
The authors would like to thank the patient and family
members who participated in this study, as well as the Royal Hospitals staff.
References
- 1. Isnard P, Rabant M, Labaye J, Antignac C, Knebelmann B, Zaidan M. Karyomegalic interstitial nephritis: a case report and review of the literature. Medicine (Baltimore) 2016 May;95(20):e3349.
- 2. Bennani Guebessi N, Karkouri M. Karyomegalic interstitial nephritis. CEN Case Rep 2016 May;5(1):23-25.
- 3. Zerres K, Mücher G, Becker J, Steinkamm C, Rudnik-Schöneborn S, Heikkilä P, et al. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet 1998 Mar;76(2):137-144.
- 4. Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJ, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers 2018 Dec;4(1):50.
- 5. Kääriäinen H. Polycystic kidney disease in children: a genetic and epidemiological study of 82 Finnish patients. J Med Genet 1987 Aug;24(8):474-481.
- 6. Zhou W, Otto EA, Cluckey A, Airik R, Hurd TW, Chaki M, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet 2012 Jul;44(8):910-915.
- 7. Seguí N, Mina LB, Lázaro C, Sanz-Pamplona R, Pons T, Navarro M, et al. Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology 2015 Sep;149(3):563-566.
- 8. National Center for Biotechnology Information. ClinVar. [cited 2023 August 15]. Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV002441339.1.
- 9. Law S, Gillmore J, Gilbertson JA, Bass P, Salama AD. Karyomegalic interstitial nephritis with a novel FAN1 gene mutation and concurrent ALECT2 amyloidosis. BMC Nephrol 2020 Feb;21(1):74.
- 10. Mohamed M, Tellez J, Bergmann C, Gale DP, Sayer JA, Olinger E. Pseudodominant Alport syndrome caused by pathogenic homozygous and compound heterozygous COL4A3 splicing variants. Ann Hum Genet 2022 May;86(3):145-152.
- 11. Ebner K, Reintjes N, Feldkötter M, Körber F, Nagel M, Dötsch J, et al. A case report on the exceptional coincidence of two inherited renal disorders: ADPKD and Alport syndrome Clin Nephrol 2017 Jul;88(1):45-51.